

Many interesting reactions take place in the liquid phase. For example, biochemical reactions occur in the aqueous environment or sometimes at the water-lipid interface. Organic synthesis is typically carried out in solvents. The observation that the environment has a significant effect on reaction outcomes was probably known to medieval alchemists. Studies by Berhelot and Pean de Saint Gilles in 1860s showed that the rates of homogeneous chemical reactions considerably depend on the environment. They noticed, for example, that both the formation and the hydrolysis of ethyl acetate was greatly retarded in the vapor phase as compared to water. Later, in 1890 Menshutkin studied the reaction between trialkylamines and haloalkanes in twenty three different solvents and reported that "solvents are by no means inert in chemical reactions". It is now widely appreciated that molecular properties, such as acid dissociation constants, are strongly solvent dependent. Rates of chemical reactions, especially when polar transition states are involved, can vary many orders of magnitude depending on the solvent. It is clear that computational description of solvent effects is crucial if quantitative agreement with experiments is being sought. In many cases, even qualitative insight requires that solvent effects are accounted for.
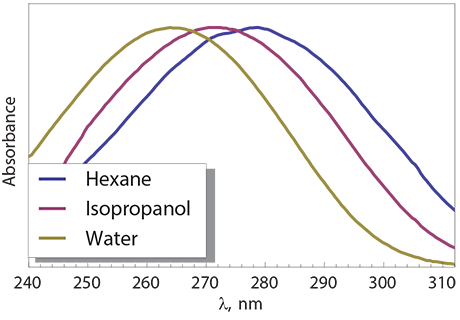 UV-Vis spectral properties of many molecules depend on the solvent. One of the best understood examples is provided by the n → π* transition in acetone. In the gas phase, this symmetry-forbidden transition is observed as a weak band of peaks near 276 nm (4.49 ev). When dissolved in water, the n → π* transition is observed at 265 nm (4.68 eV). Such changes in the spectrum with solvent are called solvatochromic shifts. When the spectral maximum shifts to longer wavelengths, we say that a red-shift occurred. In case of acetone, a spectral blue-shift occurred when acetone was moved from the gas phase to water. The solvatochromism in acetone can be understood in terms of dipolar interactions between the polar solute and the solvent. In the gas phase and non-polar solvents, the dipolar interactions are negligible both in the ground and the excited state. In water, the strength of dipolar interactions is significant. More importantly, the ground state and the excited state enjoy different stabilisation by water because the dipole moment of acetone is different in the ground and excited states.
UV-Vis spectral properties of many molecules depend on the solvent. One of the best understood examples is provided by the n → π* transition in acetone. In the gas phase, this symmetry-forbidden transition is observed as a weak band of peaks near 276 nm (4.49 ev). When dissolved in water, the n → π* transition is observed at 265 nm (4.68 eV). Such changes in the spectrum with solvent are called solvatochromic shifts. When the spectral maximum shifts to longer wavelengths, we say that a red-shift occurred. In case of acetone, a spectral blue-shift occurred when acetone was moved from the gas phase to water. The solvatochromism in acetone can be understood in terms of dipolar interactions between the polar solute and the solvent. In the gas phase and non-polar solvents, the dipolar interactions are negligible both in the ground and the excited state. In water, the strength of dipolar interactions is significant. More importantly, the ground state and the excited state enjoy different stabilisation by water because the dipole moment of acetone is different in the ground and excited states.
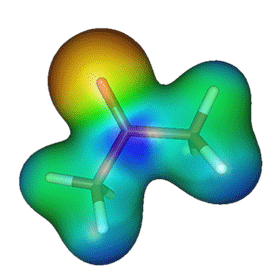 The figure on the right shows the electrostatic potential of acetone, mapped on the electron density surface. This figure illustrates a well-known fact that acetone is a polar molecule with excess negative charge localized around the oxygen atom. The simplest quantitative description of polarity of molecules is provided by its electric dipole moment. The dipole moment can be measured experimentally by studying how the microwave spectral lines of a molecule change in the presence of electric field (the Stark effect). For example, the most recent results suggest that acetone's electric dipole moment is 2.93 Debye (Dorosh & Kisiel, Acta Phys. Polonica A, 112, S-95, 2007). The dipole moment can be also obtained by analyzing the molecular charge distribution from quantum mechanical calculations. The results of such calculations depend somewhat on the level of theory and basis set used. In general, the HF method tends to overestimate dipole moments (HF/aug-cc-pVTZ prediction for acetone is 3.48 Debye) and consideration of electron correlation via MP2 improves the results notably (MP2/aug-cc-pVTZ result for acetone is 2.98). The coupled cluster methods are reliable for the estimation of dipole moments. For example, CCSD-T/aug-cc-pVTZ predicts (via numeric differentiation of energies w.r.t. field) that the dipole moment of acetone in the ground state is 2.94 Debye. The excited state has different electron distribution, and thus different dipole moment. To understand how the dipole moments of the ground and excited state differ, it is instructive to analyze the spatial localization of the orbitals involved in the n → π* transition. The images below show the highest occupied molecular orbital (HOMO), which is the non-bonding orbital, and the lowest unoccupied molecular orbital (LUMO), which is the π* orbital.
The figure on the right shows the electrostatic potential of acetone, mapped on the electron density surface. This figure illustrates a well-known fact that acetone is a polar molecule with excess negative charge localized around the oxygen atom. The simplest quantitative description of polarity of molecules is provided by its electric dipole moment. The dipole moment can be measured experimentally by studying how the microwave spectral lines of a molecule change in the presence of electric field (the Stark effect). For example, the most recent results suggest that acetone's electric dipole moment is 2.93 Debye (Dorosh & Kisiel, Acta Phys. Polonica A, 112, S-95, 2007). The dipole moment can be also obtained by analyzing the molecular charge distribution from quantum mechanical calculations. The results of such calculations depend somewhat on the level of theory and basis set used. In general, the HF method tends to overestimate dipole moments (HF/aug-cc-pVTZ prediction for acetone is 3.48 Debye) and consideration of electron correlation via MP2 improves the results notably (MP2/aug-cc-pVTZ result for acetone is 2.98). The coupled cluster methods are reliable for the estimation of dipole moments. For example, CCSD-T/aug-cc-pVTZ predicts (via numeric differentiation of energies w.r.t. field) that the dipole moment of acetone in the ground state is 2.94 Debye. The excited state has different electron distribution, and thus different dipole moment. To understand how the dipole moments of the ground and excited state differ, it is instructive to analyze the spatial localization of the orbitals involved in the n → π* transition. The images below show the highest occupied molecular orbital (HOMO), which is the non-bonding orbital, and the lowest unoccupied molecular orbital (LUMO), which is the π* orbital.
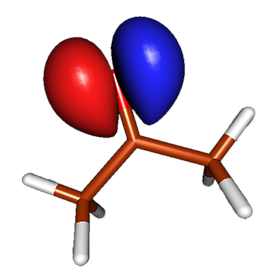
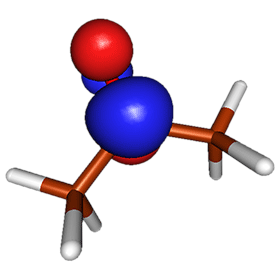
The orbital pictures reveal that the n → π* electronic transition involves transfer of one electron from a region near the oxygen atom to a region near the carbonyl carbon. This process is expected to reduce the dipole moment: the excited state of acetone is not as polar as the ground state. Our qualitative reasoning is confirmed by accurate quantum mechanical calculations: the most recent computational results suggest that the dipole moment of the n → π* excited state of acetone is 1.78 Debye (Pašteka, Melichercík, Neogrády & Urban, Mol. Phys. 2012).
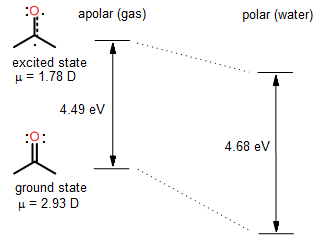 As mentioned above, the solvatochromic shifts can be rationalized by considering interactions between the solute and the solvent. A more polar ground state of acetone enjoys greater stabilization by polar water than a less polar excited state. As a result, the energy gap between the two states is greater in the presence of polar solvent in comparison with the energy gap in the gas phase or when dissolved in non-polar solvent. Larger energy gap means that light of higer frequency, or of shorter wavelength, is needed to promote the electron from the n orbital to the π* orbital. In summary, one can rationally predict the direction of the solvatochromic shift when the nature of the electronic transition and the electronic structure of the molecule are well understood. Alternatively, experimental directions and magnitudes of solvatochromic shifts are sometimes used to determine the nature of the electronic transition, and to estimate the dipole moment of the excited state.
As mentioned above, the solvatochromic shifts can be rationalized by considering interactions between the solute and the solvent. A more polar ground state of acetone enjoys greater stabilization by polar water than a less polar excited state. As a result, the energy gap between the two states is greater in the presence of polar solvent in comparison with the energy gap in the gas phase or when dissolved in non-polar solvent. Larger energy gap means that light of higer frequency, or of shorter wavelength, is needed to promote the electron from the n orbital to the π* orbital. In summary, one can rationally predict the direction of the solvatochromic shift when the nature of the electronic transition and the electronic structure of the molecule are well understood. Alternatively, experimental directions and magnitudes of solvatochromic shifts are sometimes used to determine the nature of the electronic transition, and to estimate the dipole moment of the excited state.
The solvatochromic shift in the visible region leads to the change of the color of the solution. For example, the 2-(4'-hydroxystyryl)-N-methyl-quinolinium-betaine absorbs near 585 nm in the nonpolar solvent chloroform but absorbs high-energy light at 410 nm in water. As a result, the molecule appears blue in chloroform and red in water. In the case of the betaine, chemical intuition suggests that the highly polar ground state (zwitterion!) must be well stabilized by water while the non-polar excited state enjoys less stabilization in water. The result of this differential stabilization is that the energy gap associated with the n → π transition increases as the solvent polarity increases.
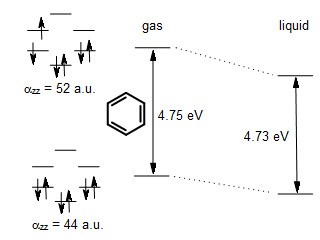 The π → π* transitions in nonpolar molecules, such as benzene or toluene, often show small spectral red-shifts when the environment is changed from the gas phase to liquid. Such non-polar molecules have zero (e.g. benzene) or very small (e.g. toluene) dipole moments, and the above-described dipolar interactions are not significant. Non-polar aromatic molecules interact with their surrounding primarily via their permanent quadrupole moment and temporarily induced dipole moments (London dispersion). In symmetric non-polar molecules such as ethylene, butadiene, benzene, toluene, or naphtalene, the excitation of electron from π to π* orbital does not change the quadrupole moment appreciably. However, the polarizability, which determines the strength of the London dispersion interaction, increases noticeably upon π → π* excitation because the electron in the π* orbital is farther away from the attractive nuclear framework. For example, the perpendicular αzz component of benzene increases from 44 atomic units to 52 atomic units upon the lowest-energy π → π* excitation (Christiansen, Hättig & Jørgensen, Spectrochimica Acta, Part A 55, 509 (1999)). Thus, the π* excited state enjoys stronger London dispersion interaction with the surrounding solvent than the less polarizable ground state, and the energy gap in the solvent is smaller than in the gas phase. In the case of benzene, the magnitude of the solvatochromic red shift is rather small.
The π → π* transitions in nonpolar molecules, such as benzene or toluene, often show small spectral red-shifts when the environment is changed from the gas phase to liquid. Such non-polar molecules have zero (e.g. benzene) or very small (e.g. toluene) dipole moments, and the above-described dipolar interactions are not significant. Non-polar aromatic molecules interact with their surrounding primarily via their permanent quadrupole moment and temporarily induced dipole moments (London dispersion). In symmetric non-polar molecules such as ethylene, butadiene, benzene, toluene, or naphtalene, the excitation of electron from π to π* orbital does not change the quadrupole moment appreciably. However, the polarizability, which determines the strength of the London dispersion interaction, increases noticeably upon π → π* excitation because the electron in the π* orbital is farther away from the attractive nuclear framework. For example, the perpendicular αzz component of benzene increases from 44 atomic units to 52 atomic units upon the lowest-energy π → π* excitation (Christiansen, Hättig & Jørgensen, Spectrochimica Acta, Part A 55, 509 (1999)). Thus, the π* excited state enjoys stronger London dispersion interaction with the surrounding solvent than the less polarizable ground state, and the energy gap in the solvent is smaller than in the gas phase. In the case of benzene, the magnitude of the solvatochromic red shift is rather small.
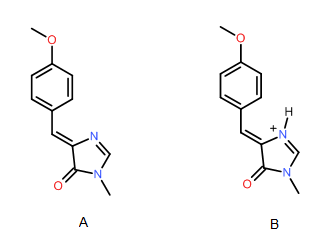 Analysis of solvatochromic shifts in molecules where both π → π* and n; → π* transitions can take place is more challenging. For example, in conjugated heteroaromatic systems, an excited state can arise via a mixture of π → π* and n; → π* transitions. Furthermore, a π → π* transition in heteroaromatic systems can alter both the dipole moment and the quadrupole moment of the system, so prediction of solvatochromic shifts becomes much more challenging. For example, the methoxy analog of the chromophore found in the green fluorescent protein shows virtually no solvatochromisms in the neutral state (A) but displays complex solvatochromism in the cationic state (B). One possible explanation for the lack of solvatochromism in the neutral form is that long-wavelength maximum is dominated here by the π → π* transition, which does not alter the dipole moment significantly. The same transition in the cationic species is a mixture of the π → π* and n → π* transitions, the latter being stabilized because transfer of the electron from carbonyl oxygen to π* orbital on carbonyl carbon is favored by the adjacent positive charge. In many cases, computational analysis of ground and exited states can reveal the direction and magnitude of solvatochromic shifts.
Analysis of solvatochromic shifts in molecules where both π → π* and n; → π* transitions can take place is more challenging. For example, in conjugated heteroaromatic systems, an excited state can arise via a mixture of π → π* and n; → π* transitions. Furthermore, a π → π* transition in heteroaromatic systems can alter both the dipole moment and the quadrupole moment of the system, so prediction of solvatochromic shifts becomes much more challenging. For example, the methoxy analog of the chromophore found in the green fluorescent protein shows virtually no solvatochromisms in the neutral state (A) but displays complex solvatochromism in the cationic state (B). One possible explanation for the lack of solvatochromism in the neutral form is that long-wavelength maximum is dominated here by the π → π* transition, which does not alter the dipole moment significantly. The same transition in the cationic species is a mixture of the π → π* and n → π* transitions, the latter being stabilized because transfer of the electron from carbonyl oxygen to π* orbital on carbonyl carbon is favored by the adjacent positive charge. In many cases, computational analysis of ground and exited states can reveal the direction and magnitude of solvatochromic shifts.
One approach to model solvent effects is to surround the solute with a small number of explicit solvent molecules during the calculation. For example, one could optimize the structure of the betaine dye in the electronic ground state by placing couple of solvent molecules near the negatively and positively charged centers; such calculation would correctly predict the direction of the solvatochromic shift. However, the magnitude of the solvent effect depends strongly on details: factors such as the number of water molecules and their placement affects results greatly. Addition of a second layer of solvent might mitigate this problem but then the number of solvent molecules is so large that typical QM calculations become unfeasible.
Alternative approach is to describe solvation by its average effect that mainly arises from dipolar interactions. In this model, the polar solute polarizes the surrounding dielectric medium. The polarized medium acts as a reaction field that interacts with the solute. The advantage of such approach is simplicity inn setting up the computations but specific interactions, such as strong hydrogen bonding with the solvent is difficult to model. Furthermore, geometry optimization in the presence of polarizable continuum is not as efficient as geometry optimization in the gas phase, and it is not uncommon that optimizations of certain geometries fail when a specific implementation of reaction field is chosen. In Gaussian, the implicit solvent model calculation is invoked via the SCRF keyword.
The input file for CIS calculation with PC GAMESS for formaldehyde in the gas phase is shown below:
$contrl scftyp=rhf cityp=cis runtyp=energy units=bohr d5=.t. icut=10 inttyp=hondo nosym=1 $end $system mwords=170 timlim=6600 $end $guess guess=huckel $end $basis extfil=.t. gbasis=acc-pvtz $end $cis nstate=6 $end $data Formaldehyde H-CHO ground state CNv 2 C 6 0.00000000E+00 0.00000000E+00 -1.00945571E+00 O 8 0.00000000E+00 0.00000000E+00 1.28258204E+00 H 1 0.00000000E+00 1.76884859E+00 -2.10196104E+00 $end
Run this by issuing pcgamess -f -i form_cis.inp -o form_cis.out -b /usr/local/pcgamess/acc-pvtz.lib. Examine the output file with a text editor and with MOLDEN to identify orbitals involved in the lowest energy forbidden transition. Then build a formaldehyde water complex; keep the structure of formaldehyde and water fixed and calculate the CIS spectrum of the complex. Interpret the results.