

The simple CIS approach is accurate in certain special cases, in particular for so-called charge transfer transitions. In the CIS approach we use orbitals of the Hartree-Fock solution to generate all singly excited determinants of the configuration interaction expansion. This treatment can be thought of as the Hartree-Fock method for excited states. It allows one to simultaneously solve for a large number of excited states and to optimize the geometry of any (desired) selected state. Both spin singlet and spin triplet states can be generated. The CIS method has some appealing features:
There are two main problem with the CIS method. First, it is appropriate only for transitions for which the ground state and the excited state are well-described by a single-configuration (such as Hartree-Fock) reference wave function. The ground state of most molecules near their equilibrium geometry is well described by a single reference (ozone is a notable exception). However, some exited states in many important chromophores (such as benzene) have a significant multi-reference character. Also, a close spacing on d-shell electron energy levels in transition metal complexes means that a single-reference description is inappropriate. In such cases, typical errors are 1 eV, which makes it difficult to assign observed spectral lines in the absence of symmetry.
Second, the CIS method, being an analog of the Hartree-Fock method for the excited states, does not include any electron correlation. This would not be a problem if the ground and excited state were stabilized by electron correlation by the same degree. This is rarely the case, and CIS typically overestimates excitation energies. The obvious solution is to describe electron correlation both in the ground and excited state but a care must be taken to provide a balanced description for all states. Some approaches, such as CISD are completely inappropriate because a closed-shell ground state would enjoy a good electron correlation while the singly excited state would not benefit as much from from virtual double excitations. Thus, CISD energies grossly overestimate the true transition energies. A partially satisfying solution is provided in the CIS-MP2 method that includes a perturbation correction for double excitations. A more balanced description is provided by the CIS(D) method which can be thought of as an analogue of MP2 for the excited states. Recent studies show that CIS(D) is quite reliable (mean error about 0.2 eV) for the description of n → π* valence shell transitions in systems where a single-reference dominates ground and excited states.
If the single reference is not adequate, multi-reference methods offer a way to calculate excitation energies. The simplest multi-reference method is CASSCF, which ignores dynamic electron correlation. More advanced approaches, such as CASPT2 and XMCQDPT add a perturbative electron correlation based on multi-reference wave function (CASSCF); such methods provide a good accuracy in many cases. The main drawback of such multi-reference methods is that their application requires a significant user input by deciding which electrons and orbitals should be included in the multi-reference treatment.
The table below compares the performance of a CIS calculation with different methods in predicting the UV spectrum of formaldehyde:
EXP CIS CIS-MP2 CIS(D) TDHF TDDFT CASSCF CASPT2 EOM-CCSD CC3 Singlet Valence Excited States 1 1A2 (n → π*) 3.79* 4.48 4.58 3.98 4.35 3.92 4.62 3.91 4.04 3.88 1 1B1 (σ → π*) 8.68 9.66 8.47 8.12 9.52 9.03 6.88 9.09 9.26 9.04 2 1A1 (π → π*) NObs 9.36 7.66 7.26 9.55 8.43 10.24 9.77 10.0 9.18 Singlet Rydberg Excited States 1 1B2 (n → 3s) 7.11 8.63 6.85 6.44 8.59 6.87 6.88 7.30 7.04 N/A 2 1A2 (n → 3p) 8.37 9.78 7.83 7.50 9.74 7.89 8.17 8.32 8.21 8.21 2 1B2 (n → 4s) 9.26 10.8 8.94 N/A N/A N/A N/A N/A 9.35 N/A Computational data sources: Gwaltney et al, Chem. Phys. Lett., 248, 189 (1996) using 6-311(2+,2+)G(d,p) basis Wiberg et al J. Phys. Chem. 106, 4192, 2002 (2008) using 6-311(2+,2+)G(d,p) basis Schreiber et al, J. Chem. Phys., 128, 134110 (2008) using bases up to d-aug-cc-pVTZ Head-Gordon et al, Chem. Phys. Lett. 219, 21 (1994) for CIS(D) Sunanda et al, Spectrosc. Lett. 45, 65 (2011) for some CIS data * The estimates for the position of 0-0 vertical transition range from 3.5 eV to 4.2 eV. This forbidden transition is seen in absorption spectra as a broad system of peaks due to many vibrational transitions. Analogous vibrational structure is also seen in the energy loss spectra from electron impact.
Note that while the CIS method generally overestimates vertical ionization energies, the n → π* transition in formaldehyde is given fairly well by the CIS, and the MP2 correction improves some results considerably. Another method similar to CIS is TDHF (time-dependent Hartree-Fock), which is a modification of CIS to include some ground state electron correlation. The last of the affordable and easy-to-use methods is the time-dependent density functional theory (TD-DFT). This methods performs well for low-lying electronic states that are not charge transfer transitions but TD-DFT fails to model excited states of linear polyenes. The last four columns refer to higher level methods that are generally more difficult to set up, or very time-consuming to run. Currently, higher level wavefunction-based methods (e.g. CASPT2 or EOM-CCSD) offer the best choice for small molecules.
The CIS method is implemented in many computer programs including Gaussian and Firefly (PC GAMESS). To run the calculation with Gaussian, specify the keyword CIS. The number of desired excited states can be specified as an option to the CIS keyword: CIS(NStates=8) requests the 8 lowest excited states. The CIS calculation is more resource-consuming than the Hartree-Fock calculation, and calculations with large basis sets, such as aug-cc-pVTZ, may not be possible for larger molecules. Smaller basis such as 6-31+G(d) may be feasible for larger molecules, but one shall worry about loss of accuracy when too small of a basis set is used. If one is only interested in valence-shell transitions, small basis sets can be used. However, for the description of diffuse Rydberg states, basis sets with multiple diffuse functions (e.g. d-aug-cc-pVTZ) are critical.
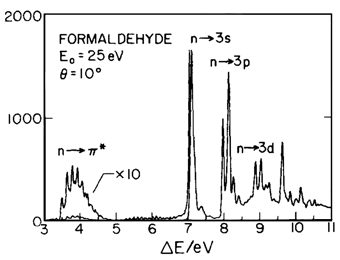 Perform a CIS calculation of formaldehyde. A sample input file for running this calculation with Gaussian is shown below. Analyze the output and try to identify some excitations that have been observed experimentally based on the excitation energies. The energy-loss from electron impact spectrum for formaldehyde in the gas phase is shown on the right (measured by Walzl, Koerting, and Kuppermann; J. Chem. Phys. 87, 3796 (1987)). Notice that this is easy for the lowest energy state but energy values alone do not allow one to assign the peaks. In symmetric molecules, the symmetry of excited states is very helpful for identification of transitions. For example, some transitions, such as n → π* are symmetry-forbidden in planar formaldehyde and can be recognized by near-zero oscillator strengths.
Perform a CIS calculation of formaldehyde. A sample input file for running this calculation with Gaussian is shown below. Analyze the output and try to identify some excitations that have been observed experimentally based on the excitation energies. The energy-loss from electron impact spectrum for formaldehyde in the gas phase is shown on the right (measured by Walzl, Koerting, and Kuppermann; J. Chem. Phys. 87, 3796 (1987)). Notice that this is easy for the lowest energy state but energy values alone do not allow one to assign the peaks. In symmetric molecules, the symmetry of excited states is very helpful for identification of transitions. For example, some transitions, such as n → π* are symmetry-forbidden in planar formaldehyde and can be recognized by near-zero oscillator strengths.
%NProc=1
%Mem=200MW
%Chk=Formald_CIS.chk
# CIS(NStates=6)/aug-cc-pVTZ MaxDisk=4GW
Formaldehyde MP2/aug-cc-pVTZ minimum for UV spectra
0 1
C
O,1,oc2
H,1,hc3,2,hco3
H,1,hc4,2,hco4,3,dih4,0
Variables:
oc2=1.21289414
hc3=1.10017879
hco3=121.7009888
hc4=1.10017879
hco4=121.7009888
dih4=180.
CIS calculations can be readily performed with larger molecules. Below is a sample output from the UV spectrum of a nucleobase uracil in Cs geometry with 6-31+G(2d,p) basis. The experimental spectrum of uracil in water shows two intense bands, centered around 257 nm and 220 nm. Based on the calculated intensities (f values are the oscillator strengths, which are the measures of intensity), these can be identified as transitions to the excited state 2 and to the excited state 8. Notice that excited state 1 has a very small intensity: this transition is nearly forbidden by orbital symmetry considerations. In this case the CIS transition energies are significantly in error. The HOMO is orbital 43; the LUMO is orbital 44. Thus, the main contribution to state 2 (as determined by squaring the given coefficient) arises from the excitation of an electron from the HOMO to LUMO + 5. You can look at the shape of orbitals involved using MOLDEN.
Excited State 1: Singlet-A" 5.7786 eV 214.56 nm f=0.0008
43 -> 44 0.57421
43 -> 45 -0.29806
43 -> 46 0.13312
43 -> 50 0.12401
This state for optimization and/or second-order correction.
Copying the CI singles density for this state as the 1-particle RhoCI density.
Excited State 2: Singlet-A' 5.8742 eV 211.06 nm f=0.3761
43 -> 47 -0.10128
43 -> 49 0.66751
Excited State 3: Singlet-A" 6.5816 eV 188.38 nm f=0.0023
43 -> 44 0.35589
43 -> 45 0.48737
43 -> 46 -0.24746
43 -> 48 -0.11210
Excited State 4: Singlet-A" 6.6584 eV 186.21 nm f=0.0002
37 -> 49 0.23353
41 -> 49 0.54352
41 -> 57 -0.20644
41 -> 72 0.10528
41 -> 78 -0.13593
Excited State 5: Singlet-A' 7.0599 eV 175.62 nm f=0.0028
43 -> 47 0.66177
43 -> 52 -0.15451
Excited State 6: Singlet-A" 7.0916 eV 174.83 nm f=0.0037
39 -> 44 0.10592
43 -> 45 0.19574
43 -> 46 0.53360
43 -> 48 -0.12071
43 -> 50 -0.23089
43 -> 51 0.16163
43 -> 55 0.13265
Excited State 7: Singlet-A" 7.5672 eV 163.84 nm f=0.0108
39 -> 44 0.10410
43 -> 45 0.15085
43 -> 46 0.15656
43 -> 48 -0.23182
43 -> 50 0.54108
43 -> 56 0.16348
43 -> 62 -0.11541
Excited State 8: Singlet-A' 7.6361 eV 162.36 nm f=0.5017
39 -> 57 -0.10651
43 -> 52 0.10904
43 -> 54 -0.28671
43 -> 57 0.50535
43 -> 59 -0.19194
43 -> 61 -0.19798
Analyze the result of CIS calculation of purine and identify molecular orbitals involved in the lowest energy transition. Then analyze the result of molecular orbital calculation of purine; visually examine the orbitals that give the largest contribution to the lowest energy excitation. Is this a π → π* or n → π* type transition?