

The structure that you built and optimized may or may not correspond to the lowest energy structure. In case of large molecules, the initial structure is likely to be different from the lowest energy conformer. For not too large flexible molecules, the lowest energy structure can be found by generating a large number of different conformations and minimizing each. Different conformers are generated by systematically or randomly altering the torsional angles in the molecule. Such analysis is commonly called the conformational analysis. Conformational analysis requires a large number of energy evaluations and is practical only using molecular mechanics force fields. In order to perform conformational analysis, the program should be able to recognize your molecule and assign suitable force field parameters. Because parameters for some molecular connectivities are missing, not all molecules can be analyzed with many common force fields.
We will be using a general-purpose molecular modeling software TINKER to perform conformational analysis of isopropanol. One of the reasons for using TINKER is that it is free and you can install TINKER on your personal Linux, Windows XP or MacOS computer. TINKER provides a suite of molecular-mechanics based modeling tools that can be executed from the Unix shell, linked to a third party program, such as molden, or launched from TINKER's own Java-based graphical user interface ffe. For example, the geometry optimization that you performed in MOLDEN in the earlier part invoked TINKER's minimize program to find a local minimum. In this part, you will learn how to run TINKER programs interactively using the Unix shell.
It is often useful to compare the three-dimensional structure of a drug candidate with the known bioactive conformer of a known ligand. One may also want to compare different conformers a molecule to see the differences between low-energy and higher energy conformers. This is often best accomplished by superimposing molecules. Molecules can be superimposed either graphically or automatically by the computer. Most commercial molecular modeling programs allow superimposing molecules. Several free programs, such as gOpenMol, PyMOL and UCFS Chimera allows users to superimpose molecules graphically. TINKER allows both automatic (via superpose program) and graphical (via the Force Field Explorer GUI) superposition of molecules. The automatic superposition in TINKER is performed using the superpose program by minimizing the RMS difference between atomic positions. You will be using UCSF Chimera in this exercise to superimpose the conformers of isopropanol.
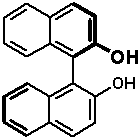 The highest point along the lowest-energy path between two stable conformers corresponds to the rotational saddle point. Knowing the energy of such saddle points is important because the hight of the rotational barrier determines how rapidly the interconversion between isomers occurs. The rotation around most single bonds is rapid because the rotational barrier is fairly low. However, in some systems the rotational barrier around single bonds is high due to steric hindrance. For example, the barrier around the rotation of the carbon-carbon single bond in 1,1-binaphthyls is so high that one can separate two optical antipodes (atropomers); such molecules serve as useful chiral catalysts or materials for chiral liqud crystals.
The highest point along the lowest-energy path between two stable conformers corresponds to the rotational saddle point. Knowing the energy of such saddle points is important because the hight of the rotational barrier determines how rapidly the interconversion between isomers occurs. The rotation around most single bonds is rapid because the rotational barrier is fairly low. However, in some systems the rotational barrier around single bonds is high due to steric hindrance. For example, the barrier around the rotation of the carbon-carbon single bond in 1,1-binaphthyls is so high that one can separate two optical antipodes (atropomers); such molecules serve as useful chiral catalysts or materials for chiral liqud crystals.
Conformational analysis algorithms can be modified such that saddle points can be optimized. The simplest approach would be to start with a structure close to the saddle point and use the Newton-Rhapson optimization method without checking for the curvature. In TINKER, the keyword SADDLEPOINT in the key file during minimization with the newton program will signal that a saddle point is being sought. Add a keyword saddlepoint to the end of the key file (e.g. isopropanol_ecl.key) with a text editor, save the file, and then run the program newton from the unix shell using the initial guess structure for the saddle point (e.g. isopropanol.xyz) as an input. Keep in mind that the Newton-Rhapson search is likely to converge to the nearest stationary point; there may be other saddle points that it will not find without your guidance. Alternatively, TINKER's program saddle can be used to locate a saddle point between the two minima.