
The chemical reactivity of molecules is largely determined by the relative energies of reactants, products, and transition states along the minimum energy path that connects reactants and products. One can quickly assess if a particular reaction is enthalpically favorable or unfavorable by minimizing the structures of the reactant and product and calculating the energy difference between the two. Similarly, one can ask if a particular mechanism is sensible by optimizing the structures of proposed intermediates and transition states along a given reaction path and asking if the energy difference between the highest energy transition state, and the reactant is consistent with the experimentally observed activation energy. If the accurately computed transition state is much higher in energy than predicted by the experiment, then the proposed mechanism is probably not correct. If the accurately computed energy difference between the transition state and the reactant matches the activation energy, one would consider the mechanism likely a correct one. If the accurately computed energy difference between the transition state and the reactant is much less than the experimental activation energy, maybe one should re-measure the experimental value!
So, if we can determine the chemical reactivity by computational studies, why wouldn't we always use computers to elucidate reaction mechanisms? The answer lies in our inability to accurately model structures and energies of large molecules in a reasonable amount of time. We can apply accurate computational methods to small molecules, but application of the same accurate methods to large molecules would make the calculation go on for too long, or would require more disk space than is available. In practice, computational chemists can calculate gas phase structures, energies, and properties of molecules consisting of a couple of heavy atoms with accuracy that rivals the best of experiments. Indeed, a good part of modern atmospheric chemistry is computational. Unfortunately, many molecules of interest to modern organic and bio-organic chemists are too large for such highly accurate methods, so approximate computational methods must be used. In practice, the following five levels of approximations are commonly made:
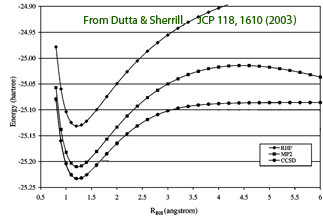 Each of these approximations comes at the cost of systematic errors. As a result, many of these methods are unsuitable for the description of bond-breaking and bond-formation reactions. Correlated methods that use a spin-restricted Hartree-Fock reference wave function often fail catastrophically at long interatomic distances. For example, the restricted MP2 method significantly overestimates the bond dissociation energy of BH at intermediate internuclear distances and the MP2 energy diverges to negative infinity as the molecule breaks apart (figure on the right). In addition to problems related to the treatment of electron correlation, the atomic orbitals in most quantum chemistry methods are represented as a sum of Gaussian functions. Calculations with fewer Gaussian functions (i.e. with small basis sets) are much faster, but using too few Gaussian functions will lead to additional errors. As a result, the casual and uncritical use of computational methods to reactions involving larger molecules is likely to yield unreliable results.
Each of these approximations comes at the cost of systematic errors. As a result, many of these methods are unsuitable for the description of bond-breaking and bond-formation reactions. Correlated methods that use a spin-restricted Hartree-Fock reference wave function often fail catastrophically at long interatomic distances. For example, the restricted MP2 method significantly overestimates the bond dissociation energy of BH at intermediate internuclear distances and the MP2 energy diverges to negative infinity as the molecule breaks apart (figure on the right). In addition to problems related to the treatment of electron correlation, the atomic orbitals in most quantum chemistry methods are represented as a sum of Gaussian functions. Calculations with fewer Gaussian functions (i.e. with small basis sets) are much faster, but using too few Gaussian functions will lead to additional errors. As a result, the casual and uncritical use of computational methods to reactions involving larger molecules is likely to yield unreliable results.
Quantum chemistry provides rigorous formulas to calculate energy of isolated molecules but the reactions of interest typically happen in the condensed medium. Organic chemists could care less about the fact that SN2 displacements of chloride by the hydroxide anion are extremely fast in the gas phase; what matters is the energetics of this reaction in the solution. Similarly, biochemists are interested how the surrounding active site residues participate in catalysis, and studying a reaction in the gas phase will reveal little about how the enzyme achieves catalysis. The incorporation of the effects of the environment into the calculation is challenging. Effects of a bulk solvent can be described via self-consistent reaction field methods. In this approach, the solvents acts as a homogenous continuous polarizable medium that attenuates electrostatic interactions between solute atoms. A promising approach for the treatment of reactions in protein active sites involves hybrid quantum mechanical/molecular mechanical (QM/MM) calculations, in which the reactive system is treated by quantum mechanics while under the influence of electrostatic and van der Waals forces from the surrounding protein atoms.
In this tutorial you will use some of the approximate quantum chemistry methods to characterize the isomerization of chorismate into prephenate. This is one of the simplest enzyme-catalyzed reactions, involving only a single substrate and no covalent intermediates. You will learn how to calculate the reaction energy (enthalpy), locate transition states, and estimate activation energies for different reaction paths. You will compare two possible mechanism of chorismate isomerization to identify the transition state with the lowest energy. This will enable you to find a suitable inhibitor for the enzyme that catalyzes this reaction.
You will calculate minimum energy geometries of chorismate and prephenate using MOLDEN and MOPAC. MOPAC is a semi-empirical quantum chemistry program. The semi-empirical approach relies on a set of atom-specific parameters, which are usually determined by minimizing the difference between the calculated and experimentally observed properties of atoms and small molecules. By using such parameterized descriptions of atoms, semi-empirical methods can provide rather simple but also accurate quantum-chemical models also for larger molecules. Semi-empirical models are important because they allow the calculation of structures and properties of very large molecules that are of interest in biochemistry and drug design. MOLDEN serves as an interface to quantum chemistry programs such as Gaussian, Gamess, and MOPAC.
First, obtain the structures of chorismate with Online SMILES Translator by National Institutes of Health. At the website, sketch the molecule as a monoanion in which the carboxylic acid connected to the six-membered ring is protonated. Make sure to specify correct stereochemistry, select MOL and 3D for output formats, and hit Translate. Save the structure (e.g. chorismate_1.mol) in your directory as a 3D MOL file. Repeat this process to generate 3D structure for prephenate.