
|
|
Enzymes are biological macromolecules that catalyze chemical reactions. They are fascinating molecules for several reasons. First, enzymes show high specificities toward their physiological substrates; compounds that differ only slightly from the natural substrate are often not acted upon. Second, activity of many enzymes can be finely tuned, allowing the metabolism proceed at a rate optimal for the well-being of the organism. Third, and maybe the most facinating aspect of enzymes action is the fact that many enzymes bring about enormous rate accelerations. For example, the enzyme rate constant kcat for decarboxylation of the amino acid arginine is 7*1019 times larger than the rate constant for the spontaneous decarboxylation of amino acids (Snider & Wolfenden, J. Am. Chem. Soc. 2000, 122,11507).
My research focuses on using computational tools for gaining better understanding on how enzymes bring about such enormous rate accelerations. Some of the factors that can contribute to catalysis and are amenable to theoretical description include:
During the past few years I have used ab initio calculations and molecular dynamics simulations to study the mechanism of several enzymes. Research projects that have been published to date include:
Globular proteins are highly flexible, conformationally dynamic macromolecules. Two types of motions can be distinguished in proteins: (i) random thermal fluctuations of atoms and groups around their average positions, and (ii) directional movements of groups or protein domains. Experimental evidence suggests that flexibility and internal motions are important for many protein functions. For example, the induced fit hypothesis postulates that the conformation of the enzyme changes during the substrate binding process such that interactions between the enzyme and the substrate are optimized. Induced fit is observed in many enzymes, such as hexokinase and lactate dehydrogenase. The role of conformational changes is also well understood in processes such as cooperative binding of oxygen to hemoglobin and allosteric regulation of glycogen phosphorylase and aspartate transcarbamoylase.
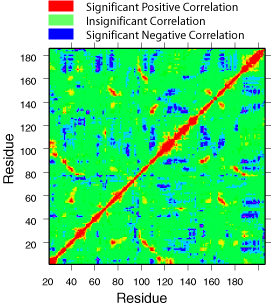 When motions in different parts of the protein occur with the same frequency, directionality, and phase, the motions are said to be correlated. Motions that occur with the same frequency and directionality, but with opposite phase are said to be anticorrelated. Correlated and anticorrelated motions in proteins are thought to play a role in catalysis, allosteric regulation, and protein folding. Very recently, experimental methods such as NMR, has allowed detection of motions with frequency that correlates with the turnover number of the enzyme. Correlated motions can be studied also by means of long molecular dynamic simulations. The correlated motions are typically displayed on a color-coded contour map such as the one shown right.
When motions in different parts of the protein occur with the same frequency, directionality, and phase, the motions are said to be correlated. Motions that occur with the same frequency and directionality, but with opposite phase are said to be anticorrelated. Correlated and anticorrelated motions in proteins are thought to play a role in catalysis, allosteric regulation, and protein folding. Very recently, experimental methods such as NMR, has allowed detection of motions with frequency that correlates with the turnover number of the enzyme. Correlated motions can be studied also by means of long molecular dynamic simulations. The correlated motions are typically displayed on a color-coded contour map such as the one shown right.
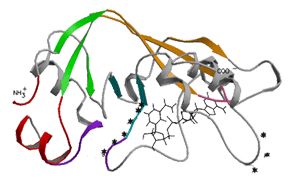 During our studies of diphtheria toxin I realized that mapping of correlated motions on the protein surface would facilitate the analysis because now the one-dimensional sequence axes of the contour plot are replaced by a three-dimensional cartoon of the actual protein structure. Such maps display all the regions of the protein that move in a concerted manner using the same color. For example, the three antiparallel strands of the β sheet move together during protein motion, and are all colored light green. Such figures reveal that major correlated motions occur within secondary structures. Interestingly, an anticorrelated motion was observed between residues (shown with stars (*)) in two loops that flank the active site.
During our studies of diphtheria toxin I realized that mapping of correlated motions on the protein surface would facilitate the analysis because now the one-dimensional sequence axes of the contour plot are replaced by a three-dimensional cartoon of the actual protein structure. Such maps display all the regions of the protein that move in a concerted manner using the same color. For example, the three antiparallel strands of the β sheet move together during protein motion, and are all colored light green. Such figures reveal that major correlated motions occur within secondary structures. Interestingly, an anticorrelated motion was observed between residues (shown with stars (*)) in two loops that flank the active site.